

For direct contact call
+31 (0)71 528 01 12
Technical Documentation
Unique Device Identifier (UDI) in the EU
What is the UDI?
The unique device identification (UDI) is a unique numeric or alphanumeric code related to a medical device. It allows for a clear and unambiguous identification of specific devices on the market and facilitates their traceability e.g. for reporting serious incidents and field safety corrective actions. The UDI comprises the following components:
- a device identifier (UDI-DI)
The UDI-DI is a fixed code and is specific to a model or a version of a device. These are codes that can be purchased at a designated entity.
- a production identifier (UDI-PI)
This part contains information on the manufacturing data, e.g., production date, expiry date, batch/lot number or a serial number.
And What is the Basic UDI-DI?
Within the EU, the manufacturer is required to assign to the devices, together with a UDI, also a Basic UDI-DI. Due to their similar names, this can be a bit confusing. The Basic UDI-DI is defined in Part C of Annex VI of the MDR. The Basic UDI-DI is the primary identifier of a device model or a generic group of devices. It is the main identifier used for records in the EUDAMED database. The Basic UDI-DI is referenced in Technical Documentation, free sale certificates, EU declarations of conformity and summary of safety and clinical performance. The Basic UDI-DI is not visible on labelling or packaging.
To generate a Basic UDI-DI, the manufacturer needs a contract with one of the UDI issuing entities designated by the European Commission (GS1, HIBCC, ICCBBA or IFA). Most of the entities have generating tools available on their website to create the basic UDI.
The Basic UDI-DI can be associated with multiple UDI-DIs (e.g. different variants) falling under the same generic group of devices but a UDI-DI shall exclusively be associated with only one Basic UDI-DI.
Where to Place the UDI (UDI-DI + UDI-PI)?
The UDI needs to be placed on the label or on the device itself and on all higher levels of device packaging, with the exception of shipping containers. The UDI carrier is required to be readable by Automatic identification and data capture (AIDC), but also by a person (HRI or Human Readable Interpretation).
For example, syringes would require a UDI per unit, but when they are sold as 10 units per box, an unique UDI would also be required for this box.
Additional Requirements
There are additional requirements to the UDI-DIs. For example , if there is a maximum number of use for a product, this number should be stated in the EUDAMED database. A change to the UDI-DI is required whenever a change to the product or otherwise occurs which may lead to misidentification of the device or ambiguity in its traceability.
Date of Application
The obligation for UDI assignment came in force on the date of application of the two new Regulations, i.e. 26 May 2021 for medical devices and 26 May 2022 for In Vitro diagnostic medical devices.
The obligation for submission of the UDI data in the EUDAMED database was fixed at 24 months after EUDAMED has become fully functional.
The obligation for placing UDI carrier on devices was fixed as follows:
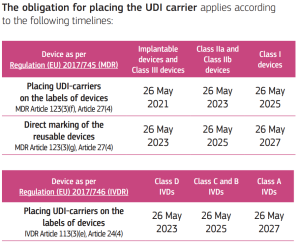
(source: https://health.ec.europa.eu/system/files/2020-09/md_faq_udi_en_0.pdf, accessed on 31-10-2022)
UDI and the QMS
The manufacturer shall assign and maintain UDI’s for its devices. To ensure a compliance with the regulations, procedures should be implemented in the manufacturers’ Quality Management System. Further guidance on this topic is provided in MDCG 2021-19.